Edwards SAPIEN 3 User manual

1
Edwards SAPIEN 3 Transcatheter Heart Valve
with the Edwards Commander Delivery System
Instructions for Use
CAUTION: Federal (USA) law restricts these devices to sale by or on the order of a physician.
Implantation of the transcatheter heart valve should be performed only by physicians who have
received Edwards Lifesciences training. The implanting physician should be experienced in
balloon aortic valvuloplasty.
Please verify that you have the latest version of the instructions for use prior to using the device
by visiting http://THVIFU.edwards.com or by calling 1.800.822.9837. In order to access the
instructions for use, an IFU Code will be required.
STERILE: The valve is supplied sterilized with glutaraldehyde solution. The delivery system,
eSheath introducer set, and crimper are supplied sterilized with ethylene oxide gas.
Edwards, Edwards Lifesciences, the stylized E logo, Carpentier-Edwards, EDWARDS COMMANDER,
Edwards eSheath, Edwards SAPIEN, Edwards SAPIEN 3, eSheath, PARTNER, PARTNER II, Qualcrimp,
SAPIEN, SAPIEN 3, TFX, and ThermaFix are trademarks of Edwards Lifesciences Corporation. All other
trademarks are the property of their respective owners.
2
1.0 Device Description
•Edwards SAPIEN 3 Transcatheter Heart Valve- Model 9600TFX (Figure 1)
The Edwards SAPIEN 3 Transcatheter Heart Valve is comprised of a balloon-expandable, radiopaque,
cobalt-chromium frame, trileaflet bovine pericardial tissue valve, and polyethylene terephthalate (PET)
fabric skirt. The leaflets are treated according to the Carpentier-Edwards ThermaFix process.
Table 1
Valve Size Height
20 mm 15.5 mm
23 mm 18 mm
26 mm 20 mm
29 mm 22.5 mm
Table 2
Native Valve Annulus Size
(TEE)
Native Valve Annulus Size
(CT) Valve Size
Area Area Derived
Diameter
16-19 mm 273 – 345 mm218.6-21 mm 20 mm
18-22 mm 338 – 430 mm2 20.7-23.4 mm 23 mm
21-25 mm 430 – 546 mm2 23.4-26.4 mm 26 mm
24-28 mm 540 – 683 mm2 26.2-29.5 mm 29 mm
Valve size recommendations are based on native valve annulus size, as measured by transesophageal
echocardiography (TEE) or computed tomography (CT). Patient anatomical factors and multiple imaging
modalities should be considered during valve size selection. Note: Risks associated with undersizing and
oversizing should be considered.
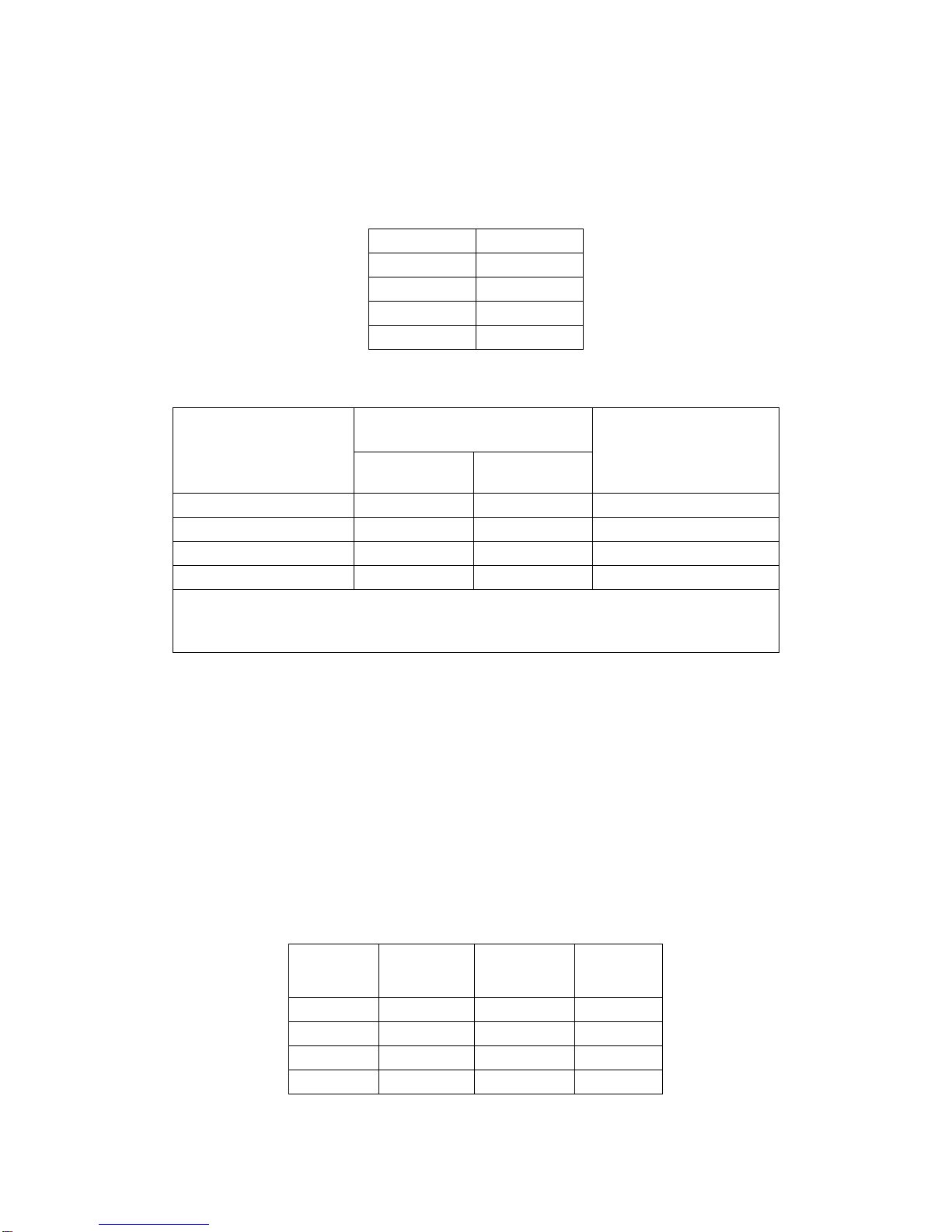
•Edwards Commander Delivery System (Figure 2)
The Edwards Commander delivery system is used for delivery of the Edwards SAPIEN 3 transcatheter
heart valve and consists of a Flex Catheter to aid in valve alignment to the balloon, tracking, and
positioning of the valve. The delivery system includes a tapered tip to facilitate crossing of the native
valve. The handle contains a Flex Wheel to control flexing of the Flex Catheter, and a Balloon Lock and
Fine Adjustment Wheel to facilitate valve alignment and positioning of the valve within the native annulus.
A stylet is included within the guidewire lumen of the delivery system. The Balloon Catheter has
radiopaque Valve Alignment Markers defining the working length of the balloon. A radiopaque Center
Marker in the balloon is provided to help with valve positioning. A radiopaque Triple Marker proximal to
the balloon indicates the Flex Catheter position during deployment. The inflation parameters for valve
deployment are:
Table 3
Model Nominal
Balloon
Diameter
Nominal
Inflation
Volume
Rated Burst
Pressure
(RBP)
9600LDS20 20 mm 11 mL 7 atm
9600LDS23 23 mm 17 mL 7 atm
9600LDS26 26 mm 23 mL 7 atm
9600LDS29 29 mm 33 mL 7 atm
3
Figure 2: Edwards Commander Delivery System
•Qualcrimp Crimping Accessory
The Qualcrimp crimping accessory (packaged with the Edwards Commander delivery system) is used
during crimping of the valve.
•Edwards eSheath Introducer Set
Refer to the Edwards eSheath Introducer Set instructions for use for device description.
•Edwards Crimper
Refer to the Edwards Crimper instructions for use for device description.
2.0 Indications
The Edwards SAPIEN 3 transcatheter heart valve, Model 9600TFX, and accessories are indicated for
relief of aortic stenosis in patients with symptomatic heart disease due to severe native calcific aortic
stenosis who are judged by a Heart Team, including a cardiac surgeon, to be at intermediate or greater
risk for open surgical therapy (i.e., predicted risk of surgical mortality ≥ 3% at 30 days, based on the
Society of Thoracic Surgeons (STS) risk score and other clinical co-morbidities unmeasured by the STS
risk calculator).
3.0 Contraindications
The valve and delivery systems are contraindicated in patients who cannot tolerate an
anticoagulation/antiplatelet regimen or who have active bacterial endocarditis or other active infections.
4.0 Warnings
•Observation of the pacing lead throughout the procedure is essential to avoid the potential risk of
pacing lead perforation.
•There may be an increased risk of stroke in transcatheter aortic valve replacement procedures, as
compared to balloon aortic valvuloplasty or other standard treatments in high or greater risk patients.
•The devices are designed, intended, and distributed for single use only. Do not resterilize or reuse
the devices. There are no data to support the sterility, nonpyrogenicity, and functionality of the
devices after reprocessing.
•Incorrect sizing of the valve may lead to paravalvular leak, migration, embolization and/or annular
rupture.
•Accelerated deterioration of the valve may occur in patients with an altered calcium metabolism.
•Prior to delivery, the valve must remain hydrated at all times and cannot be exposed to solutions
other than its shipping storage solution and sterile physiologic rinsing solution. Valve leaflets

4
mishandled or damaged during any part of the procedure will require replacement of the valve.
•Caution should be exercised in implanting a valve in patients with clinically significant coronary artery
disease.
•Patients with pre-existing mitral valve devices should be carefully assessed prior to implantation of
the valve to ensure proper valve positioning and deployment.
•Do not use the valve if the tamper evident seal is broken, the storage solution does not completely
cover the valve, the temperature indicator has been activated, the valve is damaged, or the expiration
date has elapsed.
•Do not mishandle the delivery system or use it if the packaging or any components are not sterile,
have been opened or are damaged (e.g. kinked or stretched), or the expiration date has elapsed.
•Use of excessive contrast media may lead to renal failure. Measure the patient’s creatinine level prior
to the procedure. Contrast media usage should be monitored.
•Patient injury could occur if the delivery system is not un-flexed prior to removal.
•Care should be exercised in patients with hypersensitivities to cobalt, nickel, chromium, molybdenum,
titanium, manganese, silicon, and/or polymeric materials.
•The procedure should be conducted under fluoroscopic guidance. Some fluoroscopically guided
procedures are associated with a risk of radiation injury to the skin. These injuries may be painful,
disfiguring, and long-lasting.
•Valve recipients should be maintained on anticoagulant/antiplatelet therapy, except when
contraindicated, as determined by their physician. This device has not been tested for use without
anticoagulation.
•Do not add or apply antibiotics to the storage solution, rinse solutions, or to the valve.
5.0 Precautions
•Long-term durability has not been established for the valve. Regular medical follow-up is advised to
evaluate valve performance.
•Glutaraldehyde may cause irritation of the skin, eyes, nose and throat. Avoid prolonged or repeated
exposure to, or breathing of, the solution. Use only with adequate ventilation. If skin contact occurs,
immediately flush the affected area with water; in the event of contact with eyes, seek immediate
medical attention. For more information about glutaraldehyde exposure, refer to the Material Safety
Data Sheet available from Edwards Lifesciences.
•To maintain proper valve leaflet coaptation, do not overinflate the deployment balloon.
•Appropriate antibiotic prophylaxis is recommended post-procedure in patients at risk for prosthetic
valve infection and endocarditis.
•Safety, effectiveness, and durability have not been established for valve-in-valve procedures.
•Safety and effectiveness have not been established for patients with the following
characteristics/comorbidities:
oNon-calcified aortic annulus
oSevere ventricular dysfunction with ejection fraction < 20%
oCongenital unicuspid or congenital bicuspid aortic valve
oMixed aortic valve disease (aortic stenosis and aortic regurgitation with predominant aortic
regurgitation > 3+)
oPre-existing prosthetic heart valve or prosthetic ring in any position
oSevere mitral annular calcification (MAC), severe (> 3+) mitral insufficiency, or Gorlin
syndrome

5
oBlood dyscrasias defined as: leukopenia (WBC < 3000 cells/mL), acute anemia
(Hb < 9 g/dL), thrombocytopenia (platelet count < 50,000 cells/mL), or history of
bleeding diathesis or coagulopathy
oHypertrophic cardiomyopathy with or without obstruction (HOCM)
oEchocardiographic evidence of intracardiac mass, thrombus, or vegetation
oA known hypersensitivity or contraindication to aspirin, heparin, ticlopidine (Ticlid™), or
clopidogrel (Plavix™), or sensitivity to contrast media, which cannot be adequately premedicated
oSignificant aortic disease, including abdominal aortic or thoracic aneurysm defined as maximal
luminal diameter 5 cm or greater; marked tortuosity (hyperacute bend), aortic arch atheroma
(especially if thick [> 5 mm], protruding, or ulcerated) or narrowing (especially with calcification
and surface irregularities) of the abdominal or thoracic aorta, severe “unfolding” and tortuosity of
the thoracic aorta
oAccess characteristics that would preclude safe placement of 14F or 16F Edwards eSheath
Introducer Set, such as severe obstructive calcification or severe tortuosity
oBulky calcified aortic valve leaflets in close proximity to coronary ostia
6.0 Potential Adverse Events
Potential risks associated with the overall procedure including potential access complications associated
with standard cardiac catheterization, balloon valvuloplasty, the potential risks of conscious sedation
and/or general anesthesia, and the use of angiography:
•Death
•Stroke/transient ischemic attack, clusters or neurological deficit
•Paralysis
•Permanent disability
•Respiratory insufficiency or respiratory failure
•Hemorrhage requiring transfusion or intervention
•Cardiovascular injury including perforation or dissection of vessels, ventricle, myocardium or valvular
structures that may require intervention
•Pericardial effusion or cardiac tamponade
•Embolization including air, calcific valve material or thrombus
•Infection including septicemia and endocarditis
•Heart failure
•Myocardial infarction
•Renal insufficiency or renal failure
•Conduction system defect which may require a permanent pacemaker
•Arrhythmia
•Retroperitoneal bleed
•AV fistula or pseudoaneurysm
•Reoperation
•Ischemia or nerve injury
•Restenosis
•Pulmonary edema
•Pleural effusion

6
•Bleeding
•Anemia
•Abnormal lab values (including electrolyte imbalance)
•Hypertension or hypotension
•Allergic reaction to anesthesia, contrast media, or device materials
•Hematoma
•Syncope
•Pain or changes at the access site
•Exercise intolerance or weakness
•Inflammation
•Angina
•Heart murmur
•Fever
Additional potential risks associated with the use of the valve, delivery system, and/or accessories
include:
•Cardiac arrest
•Cardiogenic shock
•Emergency cardiac surgery
•Cardiac failure or low cardiac output
•Coronary flow obstruction/transvalvular flow disturbance
•Device thrombosis requiring intervention
•Valve thrombosis
•Device embolization
•Device migration or malposition requiring intervention
•Valve deployment in unintended location
•Valve stenosis
•Structural valve deterioration (wear, fracture, calcification, leaflet tear/tearing from the stent posts,
leaflet retraction, suture line disruption of components of a prosthetic valve, thickening, stenosis)
•Device degeneration
•Paravalvular or transvalvular leak
•Valve regurgitation
•Hemolysis
•Device explants
•Nonstructural dysfunction
•Mechanical failure of delivery system, and/or accessories
•Non-emergent reoperation

7
7.0 Directions for Use
7.1 Required Equipment
Table 4:
Product Name 20 mm System
(9600CM20A) 23 mm System
(9600CM23A) 26 mm System
(9600CM26A) 29 mm System
(9600CM29A)
Model
Edwards SAPIEN 3
Transcatheter Heart
Valve
9600TFX (20 mm) 9600TFX (23 mm) 9600TFX (26 mm) 9600TFX (29 mm)
Edwards Commander
Delivery System*
9600LDS20 9600LDS23 9600LDS26 9600LDS29
Edwards eSheath
Introducer Set** 914ES 914ES 914ES 916ES
Edwards Balloon
Catheter
9350BC16 9350BC20 9350BC23 9350BC25
Inflation devices provided by Edwards Lifesciences
Edwards Crimper 9600CR
*Includes the Qualcrimp Crimping Accessory,2-piece Crimp Stopper and loader
** Or other compatible sheath provided by Edwards Lifesciences
Additional Equipment:
•20 cc syringe or larger (x2)
•50 cc syringe or larger
•High-pressure 3-way stopcock (x2)
•Standard cardiac catheterization lab equipment
•Fluoroscopy (fixed, mobile or semi-mobile fluoroscopy systems appropriate for use in percutaneous
coronary interventions)
•Transesophageal or transthoracic echocardiography capabilities
•Exchange length 0.035 inch (0.89 mm) extra-stiff guidewire
•Temporary pacemaker (PM) and pacing lead
•Sterile rinsing basins, physiological saline, heparinized saline, 15% diluted radiopaque contrast
medium
•Sterile table for valve and device preparation
7.2 Valve Handling and Preparation
Follow sterile technique during device preparation and implantation.
7.2.1Valve Rinsing Procedure
Before opening the valve jar, carefully examine for evidence of damage (e.g. a cracked jar or lid, leakage,
or broken or missing seals).
CAUTION: Valves from containers found to be damaged, leaking, without adequate sterilant, or
missing intact seals must not be used for implantation.

8
Step Procedure
1 Set up two (2) sterile bowls with at least 500 mL of sterile physiological saline to thoroughly rinse the
glutaraldehyde sterilant from the valve.
2 Carefully remove the valve/holder assembly from the jar without touching the tissue. Verify the valve
serial identification number with the number on the jar lid and record in the patient information
documents. Inspect the valve for any signs of damage to the frame or tissue.
3
Rinse the valve as follows: Place the valve in the first bowl of sterile, physiological saline. Be sure
the saline solution completely covers the valve and holder. With the valve and holder submerged,
slowly agitate (to gently swirl the valve and holder) back and forth for a minimum of 1 minute.
Transfer the valve and holder to the second rinsing bowl of sterile physiological saline and gently
agitate for at least one more minute. Ensure the rinse solution in the first bowl is not used. The
valve should be left in the final rinse solution until needed to prevent the tissue from drying.
CAUTION: Do not allow the valve to come into contact with the bottom or sides of the rinse bowl
during agitation or swirling in the rinse solution. Direct contact between the identification tag and
valve is also to be avoided during the rinse procedure. No other objects should be placed in the
rinse bowls. The valve should be kept hydrated to prevent the tissue from drying.
7.2.2Prepare the Components
Refer to the Edwards eSheath Introducer Set, Edwards Crimper and Edwards Balloon Catheter
instructions for use for device preparation.
Step Procedure
1
Visually inspect all components for damage. Ensure the Edwards Commander delivery system is fully
unflexed and the balloon catheter is fully advanced in the flex catheter.
WARNING: To prevent possible damage to the balloon shaft, ensure that the proximal end of the
balloon shaft is not subjected to bending.
2 Flush the flex catheter.
3 Carefully remove the distal balloon cover from the delivery system.
4
Remove the stylet from the distal end of the guidewire lumen and set aside. Flush the guidewire
lumen with heparinized saline and insert the stylet back into the distal end of the guidewire lumen.
Note: Failure to insert the stylet back into the guidewire lumen may result in damage to the lumen
during crimping process.
5
Place the delivery system into the default position and make sure that the flex catheter tip is covered
by the proximal balloon cover. Unscrew the loader cap from the loader tube and flush the loader cap.
Place the loader cap over the proximal balloon cover and onto the flex catheter with the inside of the
cap oriented towards the distal tip.
6 Fully advance the balloon catheter in the flex catheter.
Peel off the proximal balloon cover over the blue section of the balloon shaft.
7 Attach a 3-way stopcock to the balloon inflation port. Partially fill a 50 cc or larger syringe with
15-20 mL diluted contrast medium and attach to the 3-way stopcock.
8 Fill the inflation device provided by Edwards Lifesciences with excess volume relative to the indicated
inflation volume. Lock the inflation device and attach to the 3-way stopcock.
9 Close the 3-way stopcock to the Inflation device provided by Edwards Lifesciences and de-air the
system using the 50 cc or larger syringe. Slowly release the plunger and leave zero-pressure in the
system.
10
Close the stopcock to the delivery system. By rotating the knob of the inflation device provided by
Edwards Lifesciences, transfer the contrast medium into the syringe to achieve the appropriate
volume required to deploy the valve.

9
Step Procedure
11
Close the stopcock to the 50 cc or larger syringe. Remove the syringe. Verify that the inflation volume
is correct and lock the Inflation device provided by Edwards Lifesciences.
CAUTION: Maintain the Inflation device provided by Edwards Lifesciences in the locked
position until valve deployment.
7.2.3Mount and Crimp the Valve on the Delivery System
Step Procedure
1 Set up two (2) additional sterile bowls with at least 100 mL of sterile physiological saline to thoroughly
rinse the Qualcrimp crimping accessory.
2 Completely submerge the Qualcrimp crimping accessory in the first bowl and gently compress it to
ensure complete saline absorption. Slowly swirl the Qualcrimp crimping accessory for a minimum of
1 minute. Repeat this process in the second bowl.
3 Remove the valve from the holder and remove the ID tag.
4 Attach the 2-piece crimp stopper to the base of the crimper and click into place.
5 With the crimper in the open position, gently place the valve into the crimper aperture. Gradually
crimp the valve until it fits into the Qualcrimp crimping accessory.
6 Place the Qualcrimp crimping accessory over the valve making sure the valve is parallel to the edge
of the Qualcrimp crimping accessory.
7 Place the valve and Qualcrimp crimping accessory in crimper aperture. Insert the delivery system
coaxially within the valve on the Valve Crimp Section (2-3 mm distal to the balloon shaft) with the
inflow (outer skirt) end of the valve towards the distal end of the delivery system.
8 Crimp the valve until it reaches the Qualcrimp Stop located on the 2-piece Crimp Stopper.
9 Gently remove the Qualcrimp crimping accessory from the valve. Remove the Qualcrimp Stop from
the Final Stop, leaving the Final Stop in place.
10 Fully crimp the valve until it reaches the Final Stop.
NOTE: Ensure that the Valve Crimp Section remains coaxial within the valve.
11 Repeat the full crimp of the valve two more times for a total of three full crimps.
12 Pull the balloon shaft and lock in the default position.
13
Flush the loader with heparinized saline. Immediately advance the valve into the loader until the
tapered tip of the delivery system is exposed.
CAUTION: To prevent possible leaflet damage, the valve should not remain fully crimped
and/or in the loader for over 15 minutes.
14
Attach the loader cap to the loader, re-flush the delivery system through the flush port and close the
stopcock to the delivery system.
Remove the stylet and flush the guidewire lumen of the delivery system.
CAUTION: Keep the valve hydrated until ready for implantation.
CAUTION: The physician must verify correct orientation of the valve prior to its implantation;
its inflow (outer skirt) end should be oriented distally towards the tapered tip.
7.3 Valvuloplasty and Valve Delivery
Valvuloplasty and valve delivery should be performed under conscious sedation and/or general
anesthesia with hemodynamic monitoring in a catheterization lab/hybrid operating room with fluoroscopic
and echocardiographic imaging capabilities.
Administer heparin to maintain the ACT at ≥ 250 sec during the procedure.
CAUTION: Use of excessive contrast media may lead to renal failure. Measure the patient’s
creatinine level prior to the procedure. Contrast media usage should be monitored.
CAUTION: Procedure may require an arterial cut-down with surgical closure of the puncture site
due to the size of the arteriotomy.

10
7.3.1Baseline Parameters
Step Procedure
1 Perform a supra-aortic angiogram with fluoroscopic view perpendicular to the aortic valve.
2 Evaluate the distance of the left and right coronary ostia from the aortic annulus in relation to the valve
frame height.
3 Introduce a pacemaker (PM) lead until its distal end is positioned in the right ventricle.
4 Set the stimulation parameters to obtain 1:1 capture, and test pacing.
7.3.2Valvuloplasty
Refer to Edwards Balloon Catheter Instructions for Use (IFU) for information on device
preparation and handling.
Note: Rapid ventricular pacing should be performed when using the Edwards Balloon Catheter for
valvuloplasty prior to aortic transcatheter valve implantation.
After placement of the balloon at the intended site, begin rapid ventricular pacing. Once the systolic blood
pressure has decreased to 50 mmHg or below, balloon inflation can commence.
CAUTION: Valve implantation should not be carried out if the balloon cannot be fully inflated
during valvuloplasty.
7.3.3Valve Delivery
Step Procedure
1 Prepare and insert the Edwards eSheath Introducer Set. Refer to the Edwards eSheath Introducer
Set IFU for information on device preparation and handling.
2 Insert the loader into the sheath until the loader stops.
3
Advance the Edwards Commander delivery system, with the Edwards logo facing up, through the
sheath until the valve exits the sheath. Retract the loader to the proximal end of the delivery system.
NOTE: Maintain the proper orientation of the flex catheter (with the Edwards logo facing up)
throughout the procedure.
CAUTION: If accessing femorally or via the iliac, the valve should not be advanced through
the sheath if the sheath tip is not past the aortic bifurcation.
CAUTION: To prevent possible leaflet damage, the valve should not remain in the sheath for
over 5 minutes.
4
In a straight section of the aorta, initiate valve alignment by disengaging the Balloon Lock and pulling
the balloon catheter straight back until part of the Warning Marker is visible. Do not pull past the
Warning Marker.
WARNING: To prevent possible damage to the balloon shaft, ensure that the proximal end of
the balloon shaft is not subjected to bending.
Engage the Balloon Lock.
Use the Fine Adjustment Wheel to position the valve between the valve alignment markers.
CAUTION: Do not turn the Fine Adjustment Wheel if the Balloon Lock is not engaged.
WARNING: Do not position the valve past the distal Valve Alignment Marker. This will prevent
proper valve deployment.
CAUTION: Maintain guidewire position in the left ventricle during valve alignment.
5 Advance the catheter and use the flex wheel, if needed, and cross the aortic valve.
NOTE: Verify the Edwards logo is facing up. The delivery system articulates in a direction
opposite from the flush port.
6 If additional working length is needed, remove the loader by unscrewing the loader cap and peeling
the loader tubing from the delivery system.

11
Step Procedure
7
Disengage the Balloon Lock and retract the tip of the Flex Catheter to the center of the Triple Marker.
Engage the Balloon Lock.
8 Verify the correct position of the valve with respect to the aortic annulus.
9 As necessary, utilize the Flex Wheel to adjust the co-axiality of the valve and the Fine Adjustment
Wheel to adjust the position of the valve.
10 Before deployment, ensure that the valve is correctly positioned between the Valve Alignment
Markers and the Flex Catheter tip is over the Triple Marker.
11
Begin valve deployment:
•Unlock the Inflation device provided by Edwards Lifesciences.
•Begin rapid pacing; once systolic
blood pressure has decreased to 50 mmHg or below, balloon
inflation can commence.
•Deploy the valve by inflating the balloon with the entire volume in the Inflation device provided by
Edwards Lifesciences, hold for 3 seconds and confirm that the barrel of the inflation device is
empty to ensure complete inflation of the balloon.
•Deflate the balloon. When the balloon catheter has been completely deflated, turn off the
pacemaker.
7.3.4System Removal
Step Procedure
1 Unflex the delivery system while retracting the device, if needed. Verify that the Flex Catheter tip is
locked over the Triple Marker and remove the delivery system from the sheath.
CAUTION: Patient injury could occur if the delivery system is not unflexed prior to removal.
2 Remove all devices when the ACT level is appropriate. Refer to the Edwards eSheath Introducer Set
instructions for use for device removal.
3 Close the access site.
8.0 How Supplied
STERILE: The valve is supplied sterilized with glutaraldehyde solution. The delivery system is supplied
sterilized with ethylene oxide gas.
8.1 Storage
The valve must be stored at 10 °C to 25 °C (50 °F to 77 °F). Each jar is shipped in an enclosure
containing a temperature indicator to detect exposure of the valve to extreme temperature.
The delivery system should be stored in a cool, dry place.
9.0 MR Safety
MR Conditional
Non-clinical testing has demonstrated that the Edwards SAPIEN 3 transcatheter heart valve is MR
Conditional. A patient with this device can be scanned safely, immediately after placement of this device
under the following conditions:
•Static magnetic field of 1.5 tesla or 3 tesla
•Maximum spatial gradient field of 2500 gauss/cm (25 T/m) or less
•Maximum MR system reported, whole body averaged specific absorption rate (SAR) of 2 W/kg
(Normal Operating Mode)
Under the scan conditions defined above, the SAPIEN 3 transcatheter heart valve is expected to produce
a maximum temperature rise of 3.0 ºC after 15 minutes of continuous scanning.

12
In non-clinical testing, the image artifact caused by the device extends as far as 14.5 mm from the implant
for spin echo images and 30 mm for gradient echo images when scanned in a 3.0T MRI system. The
artifact obscures the device lumen in gradient echo images.
The implant has not been evaluated in MR systems other than 1.5 or 3.0T.
10.0 Patient Information
Patient education brochures are provided to each site and should be given to the patient to inform them of
the risks and benefits of the procedure and alternatives in adequate time before the procedure to be read
and discussed with their physician. A copy of this brochure may also be obtained from Edwards
Lifesciences by calling 1.800.822.9837. A patient implant card request form is provided with each
transcatheter heart valve. After implantation, all requested information should be completed on this form.
The serial number may be found on the package and on the identification tag attached to the transcatheter
heart valve. The original form should be returned to the Edwards Lifesciences address indicated on the form
and upon receipt, Edwards Lifesciences will provide an identification card to the patient.
11.0 Recovered Valve and Device Disposal
The explanted valve should be placed into a suitable histological fixative such as 10% formalin or 2%
glutaraldehyde and returned to the company. Refrigeration is not necessary under these circumstances.
Contact Edwards Lifesciences to request an Explant Kit.
Used delivery system may be disposed of in the same manner that hospital waste and biohazardous
materials are handled. There are no special risks related to the disposal of these devices.
12.0 Clinical Studies
SUMMARY OF PRIMARY CLINICAL STUDY
The PARTNER II Trial Overview, SAPIEN 3 Valve
SAPIEN 3 High Risk and Inoperable Cohort: The SAPIEN 3 High Risk and Inoperable Cohort of the
PARTNER II trial (PIIS3HR) was a single arm, non-randomized, historical-controlled study to compare the
third generation Edwards SAPIEN 3 valve system with the first generation Edwards SAPIEN valve system
in patients who either have high risk for surgery or cannot undergo surgery (inoperable). The valve sizes
used in the PIIS3HR trial included only the 23, 26 and 29 mm sizes. The 20 mm valve size was
introduced into the trial after enrollment was completed with the three larger sizes, thus a separate nested
registry, NR7, with identical inclusion/exclusion criteria as the PIIS3HR Cohort except for the aortic
annulus diameter, was created to collect data for the 20 mm valve. Data from the PIIS3HR cohort and
NR7 are pooled for the statistical analyses. For convenience, this combined cohort is referred to as
“PIIS3HR” hereafter.
The database included 583 eligible patients enrolled at 29 investigational sites in the U.S.
The study used an independent Data Safety Monitoring Board (DSMB) that was instructed to notify
Edwards Lifesciences of any safety or compliance issues, a Clinical Events Committee (CEC) that was
responsible for adjudicating endpoint related events reported during the trial per a priori established
VARC 2 definitions[1], an ECG core laboratory for independent analysis of rhythm, and an
echocardiographic core laboratory for independently analyzing all echocardiograms.
SAPIEN 3 Intermediate Risk Cohort: The PIIS3i Cohort of the PARTNER II trial was a single arm, non-
randomized, historical-controlled study to compare TAVR with the Edwards SAPIEN 3 valve system to the
surgical aortic valve replacement (SAVR) arm from the previous PARTNER II trial Cohort A (PIIA-SAVR)
in patients who were judged by a heart team to be at intermediate risk for open surgical therapy. The
valve sizes used in the PIIS3i study included the 20, 23, 26, and 29 mm sizes.
Patients in PIIS3i were treated between February 2014 and September 2014. Patients in PIIA-SAVR
were treated between January 2012 and November 2013. The database reflected data collected through
December 10, 2015 and included 1,078 patients in PIIS3i enrolled at 51 investigational sites in the U.S
and 1,021 patients in PIIA-SAVR enrolled at 57 investigational sites in the U.S.

13
The PIIS3i study used an independent Data Safety Monitoring Board (DSMB) that was instructed to notify
Edwards Lifesciences of any safety or compliance issues and a Clinical Events Committee (CEC) that
was responsible for adjudicating endpoint related events reported during the trial in accordance with the
pre-specified, primarily Valve Academic Research Consortium-2 VARC-2 definitions[1], with the following
exceptions:
•Prosthetic valve dysfunction was adjudicated per VARC-1
•Aortic valve reintervention was adjudicated per protocol definition
•Rehospitalization for symptoms of aortic stenosis and/or complications of the valve procedure
were adjudicated using the protocol and VARC-2 definitions as guidelines
The events in the PIIA-SAVR cohort were adjudicated by the CEC in accordance with the pre-specified,
primarily VARC-1 definitions, with the following exceptions:
•Acute Kidney Injury (AKI) was adjudicated with a modified VARC-1 definition in which the CEC
applied the 72-hour staging window to any AKI event that occurred within 30-days
•Aortic valve reintervention were adjudicated per the protocol definition
•Rehospitalization for symptoms of AS and/or complications of the valve procedure were
adjudicated using the protocol and VARC-1 as guidelines
•Bleeding events were adjudicated irrespective of whether there was an identifiable, overt source
of bleeding
An electrocardiogram (ECG) core laboratory was used for independent analysis of rhythm, an
echocardiographic core laboratory for echocardiograms, and a computerized tomography (CT) core
laboratory for baseline CTs for annulus dimensions.
PARTNER II SAPIEN 3 HIGH-RISK/INOPERABLE COHORT
Accountability
All 583 eligible patients were successfully implanted with a SAPIEN 3 valve, which constitutes the Valve
Implant (VI) population. Among the VI population, 491 patients were implanted via the transfemoral (TF)
access route, and 92 patients via the transapical (TA) or transaortic (TAo) access route.
Table 5:
Patient Accountability
SAPIEN 3
Valve
Overall
SAPIEN 3
Valve
Transfemoral
Access
SAPIEN 3
Valve
Non-
Transfemoral
Access
Eligible Patient
Population (EPP)
583 491 92
Valve Implant
(VI) Population
583 491 92
Eligible Patient Population (EPP) consists of all enrolled patients who received
treatment assignment from the database and entered into the catheterization
laboratory/hybrid suite and who remained eligible to receive the implant.
Valve Implant (VI) Population consists of all enrolled patients who received a
SAPIEN 3 valve, and retained the valve upon leaving the catheterization
laboratory/hybrid suite.
Study Population Demographics and Baseline Parameters
The demographics of the study population are summarized in Table 6, which are typical of a TAVR study
performed in the U.S.

14
Table 6:
Patient Demographics and Baseline Characteristics –
PIIS3HR VI Population
Characteristic
SAPIEN 3
Valve
Overall
(N= 583)
SAPIEN 3
Valve
Transfemoral
Access
(N= 491)
SAPIEN 3
Valve
Non-
Transfemoral
Access
(N= 92)
Age, yr
82.6 ± 8.1
82.8 ± 8.2
81.7 ± 7.5
Male sex, no. (%)
338 (58.0%)
277 (56.4%)
61 (66.3%)
STS score
8.6 ± 3.7
8.4 ± 3.5
10.0 ± 4.3
New York Heart Association (NYHA) class, no. (%):
I/II
58 (9.9%)
51 (10.4%)
7 (7.6%)
III/IV
525 (90.1%)
440 (89.6%)
85 (92.4%)
Coronary artery disease, no. (%)
444 (76.2%)
360 (73.3%)
84 (91.3%)
Previous myocardial infarction, no. (%)
117 (20.1%)
87 (17.7%)
30 (32.6%)
Previous intervention, no. (%)
Coronary-artery bypass grafting (CABG)
193 (33.1%)
145 (29.5%)
48 (52.2%)
Percutaneous coronary intervention (PCI)
199 (34.1%)
163 (33.2%)
36 (39.1%)
Prior aortic valvuloplasty
62 (10.6%)
49 (10.0%)
13 (14.1%)
Cerebral vascular accident (CVA), no. (%)
64 (11.0%)
53 (10.8%)
11 (12.0%)
Peripheral vascular disease, no. (%)
205 (35.2%)
155 (31.6%)
50 (54.3%)
Chronic obstructive pulmonary disease (COPD),
no. (%):
Any
259 (44.6%)
216 (44.1%)
43 (47.3%)
Oxygen-dependent
68 (11.8%)
58 (11.9%)
10 (11.0%)
Atrial fibrillation, no. (%)
255 (43.7%)
212 (43.2%)
43 (46.7%)
Permanent pacemaker, no. (%)
95 (16.3%)
78 (15.9%)
17 (18.5%)
Severe pulmonary hypertension, no. (%)
30 (5.1%)
24 (4.9%)
6 (6.5%)
Frailty, no. (%)
180 (30.9%)
162 (33.0%)
18 (19.6%)
Chest deformities that preclude an open chest
procedure, no. (%)
4 (0.7%)
3 (0.6%)
1 (1.1%)
Cirrhosis, no. (%)
11 (1.9%)
9 (1.8%)
2 (2.2%)
Echocardiographic findings
Effective Orifice Area (EOA), cm2
0.7 ± 0.2
0.7 ± 0.2
0.7 ± 0.1
Mean aortic-valve gradient, mmHg
45.5 ± 14.3
45.7 ± 14.4
44.0 ± 13.2
Mean left ventricular ejection fraction (LVEF), %
56.4 ± 14.8
57.0 ± 14.5
53.2 ± 15.9
Moderate or severe mitral regurgitation, no./total
no. (%)
69/541
(12.8%)
63/461
(13.7%)
6/80
(7.5%)
Safety and Effectiveness Results
Primary Endpoint
The composite rate of all-cause mortality, all stroke, and AI ≥ moderate at 30 days was 6.7% in the
SAPIEN 3 cohort and 15.6% in the SAPIEN cohort, as shown in Table 7. The resulting proportion
difference in the average treatment effect on the treated (ATT; [2]) was -6.9% (90% CI: [-13.3%, -0.5%]).
Since the upper limit of the CI was < 7.5%, the non–inferiority was met.

15
Table 7:
Primary Endpoint Analysis –
Non-Inferiority Test SAPIEN 3 Valve (PIIS3HR VI Population) vs. SAPIEN Valve
Event at 30 days SAPIEN 3
Valve
(N = 583)
SAPIEN
Valve
(N = 326)
Weighted Proportion
Difference in Average
Treatment Effect
on the Treated (ATT)
Composite of Death, Stroke
and AI ≥ Moderate)
6.7%
[5.1%, 8.6%]
1
15.6%
[12.6%, 19.5%]
1
-6.9%
[-13.3%, -0.5%]
2
1For each individual study, the two-sided 90% stratified Wilson confidence interval was provided.
2The Wald-type two-sided 90% confidence interval using weighted mean and SD is provided
The Kaplan-Meier (K-M) estimates for all-cause mortality, cardiac mortality, and all stroke at 30 days for
the SAPIEN 3 cohort and the SAPIEN cohort are provided in Table 8.
Table 8:
Death and Stroke at 30 Days –
SAPIEN 3 Valve vs. SAPIEN Valve (VI Population)
SAPIEN 3 Valve
(N = 583)
SAPIEN Valve
(N = 326)
Event at 30
Days No.
Events
No. Pts with
Events
K-M Estimated
Event Rate1
(95% CI)
No.
Events
No. Pts with
Events K-
M Estimated Event
Rate (95% CI)
Death 13 13
2.2%
([1.3%, 3.8%])
15 15
4.6%
([2.8%, 7.5%])
Cardiac
Death
8 8
1.4%
([0.7%, 2.7%])
10 10
3.1%
([1.7%, 5.7%])
All Stroke 9 9
1.6%
([0.8%, 3.0%])
14 14
4.3%
([2.6%, 7.2%])
1Kaplan-Meier (K-M) estimates at 30 days used time to first event for each patient. Events occurring after 30 days
were not included in this analysis.
Secondary Endpoints
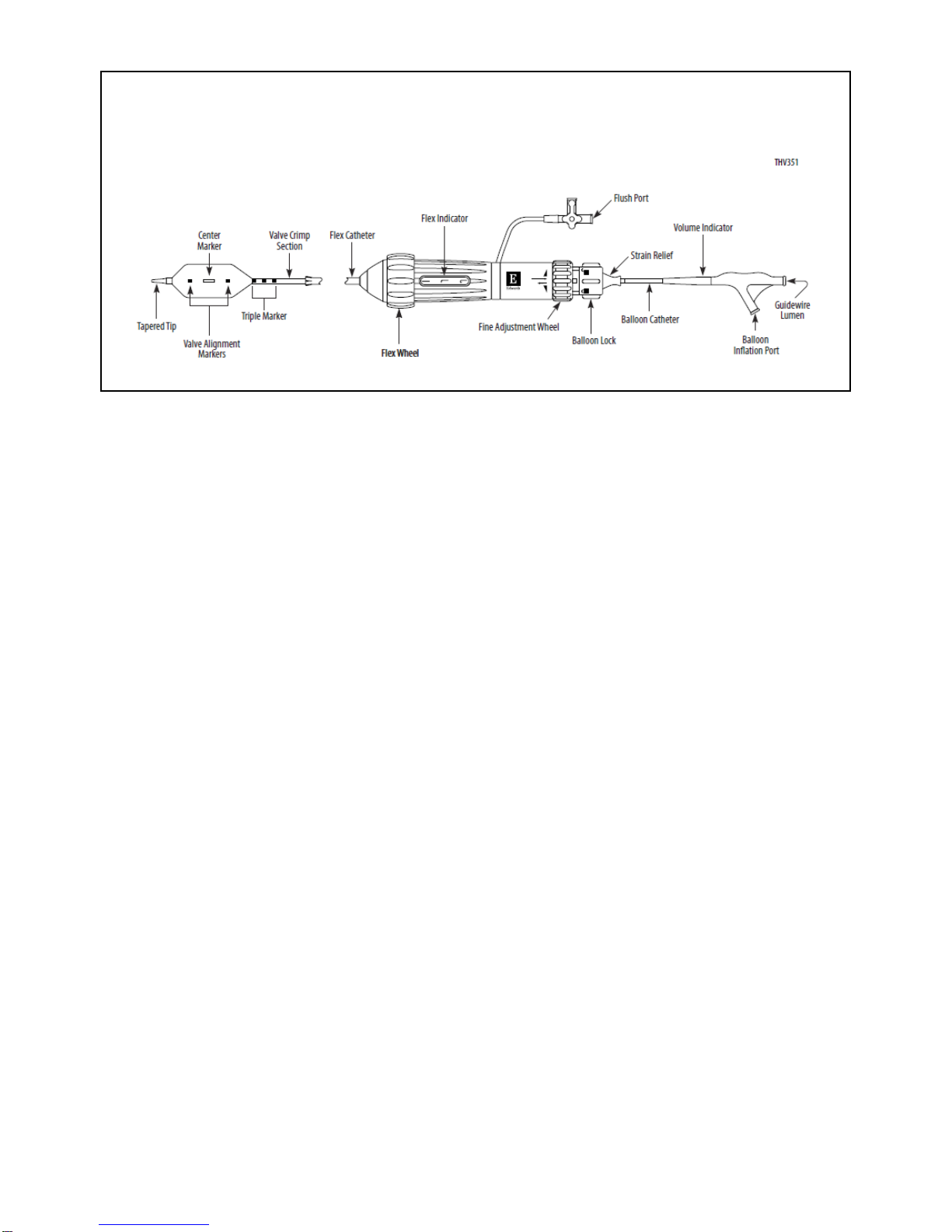
Aortic insufficiency by visit is provided in Figure 3.
16
Figure 3:
Aortic Insufficiency by Visit –
SAPIEN 3 Valve (PIIS3HR VI Population) vs. SAPIEN Valve
The proportion of patients with AI ≥ moderate at 30 days was 3.0% in the SAPIEN 3 cohort and 14.3% in
the SAPIEN cohort, which were found to be statistically significantly different (p=0.0051; Table 9).
Table 9:
Aortic Insufficiency at 30 Days
(SAPIEN 3 Valve vs. SAPIEN Valve VI Population)
Event at 30 Days SAPIEN 3
Valve
(N = 583)
SAPIEN Valve
(N = 326 )
Weighted Proportion
Difference in Average
Treatment Effect on the
Treated (ATT)
P-value
AI ≥Moderate, n/Total
no. (%) [95% CI] 16/532
(3.0%)
[1.7%, 4.8%]1
40/280
(14.3%)
[10.4%,
18.9%]1
-13.1%
[-22.2%, -3.9%]20.0051
195% Clopper-Pearson Exact confidence interval.
2The Wald-type two-sided 90% confidence interval using weighted mean and SD is provided
The rate of major vascular complications at 30 days post implantation is shown in Figure 4. The rate was
5.0% for the SAPIEN 3 cohort and 10.1% for the SAPIEN cohort, which were found to be not statistically
significantly different (p=0.0578; Table 10).

17
Figure 4:
Major Vascular Complications at 30 Days –
SAPIEN 3 Valve vs. SAPIEN Valve (VI Population)
Table 10:
Major Vascular Complications at 30 Days –
SAPIEN 3 Valve vs. SAPIEN Valve (VI Population)
Event at 30 Day SAPIEN 3
Valve
(N = 583)
SAPIEN
Valve
(N = 326)
Weighted
Proportion
Difference in
Average T
reatment
Effect on the
Treated (ATT)
P-value
Major Vascular
Complications, n/Total no.
(%) [95% CI]
29/583
(5.0%)
[3.4% , 7.1%]
33/326
(10.1%)
[7.1% , 13.9%]
1
-8.0%
[-16.2%, 0.3%]20.0578
195% Clopper-Pearson Exact confidence interval.
2
The Wald-type two-sided 90% confidence interval using weighted mean and SD is provided.
Table 11lists the hypothesis testing of the two secondary endpoints conducted with p-values in
descending order for the Hochberg multiplicity adjustment steps. The largest p-value (p=0.0578 from
major vascular complications) was greater than 0.05. As such, the null hypothesis was not rejected for the
testing of major vascular complications at 30 days. The subsequent testing of AI ≥ moderate at 30 days
had a p-value of 0.0051, which was less than 0.025. As such, the null hypothesis was rejected for
AI ≥ moderate at 30 days, indicating that the SAPIEN 3 cohort was superior over the SAPIEN cohort in
regards to AI ≥ moderate at 30 days.

18
Table 11:
Secondary Endpoints for Labeling –
SAPIEN 3 Valve vs. SAPIEN Valve (VI Population)
Endpoints
Original
p-value
Inference
Major Vascular
Complications at 30
Days
0.0578
> 0.05; reject the alternative
hypothesis. Proceed to the rest of
testing
AI at 30 Days
0.0051
< 0.025; claim superiority
Adverse Events
The key CEC adjudicated adverse events at 30 days are presented in Table 12.
Table 12:
CEC Adjudicated Adverse Events at 30 Days
(PIIS3HR VI Population)
30 Day Adverse Events SAPIEN 3 Valve
Overall
SAPIEN 3 Valve
Transfemoral
Access
TF
SAPIEN 3
Valve
Non-
Transfemoral
Access
Composite Event Rate of Death, All Stroke and
AI ≥ Moderate, n/N (%)
37/545 (6.8 %) 27/463 (5.8 %) 10/82 (12.2 %)
Death
From Any cause, n/N (%) 13/583 (2.2%) 8/491 (1.6%) 5/92 (5.4%)
From cardiovascular cause, n/N (%) 8/583 (1.4%) 5/491 (1.0%) 3/92 (3.3%)
Stroke, n/N (%) 9/583 (1.5%) 8/491 (1.6%) 1/92 (1.1%)
AI ≥ moderate, n/N (%) 16/532 (3.0%) 12/455 (2.6%) 4 /77 (5.2%)
Myocardial Infarction, n/N (%) 3/583 (0.5%) 2/491 (0.4%) 1/92 (1.1%)
Major Vascular Complications, n/N (%) 29/583 (5.0%) 26/491 (5.3%) 3/92 (3.3%)
Acute Kidney Injury, Stage III, n/N (%) 6/583 (1.0%) 4/491 (0.8%) 2/92 (2.2%)
Disabling Bleeding Event, n/N (%) 37/583 (6.3%) 27/491 (5.5%) 10/92 (10.9%)
Aortic Valve Re-Intervention, n/N (%) 6/583 (1.0%) 4/491 (0.8%) 2/92 (2.2%)
Endocarditis, n/N (%) 1/583 (0.2%) 1/491 (0.2%) 0/92 (0.0%)
Conduction Disturbance Requiring Permanent
Pacemaker, n/N (%) 76/583 (13.0%) 65/ 491 (13.2%) 11/ 92 (12.0%)
Other Results
Procedural Information
Overall, the mean duration in the catheterization laboratory/hybrid suite was 192.8 ± 59.3 min, the mean
total procedure time was 86.3 ± 44.2 min, and the mean total anesthesia time was 193.7 ± 62.9 min.
These duration times were slightly shorter in the TF patients. General anesthesia was used in the vast
majority of cases; 15.9% of the TF patients had conscious sedation. Correct positioning of the valve was
achieved in 99.1% of the patients. Five patients (0.9%; including 3 TF patients) were implanted with a
second valve. One patient (0.2%) experienced valve embolization following rupture of the delivery balloon
on annular calcium. This patient was converted to surgical aortic valve replacement and later died from
aortic dissection.

19
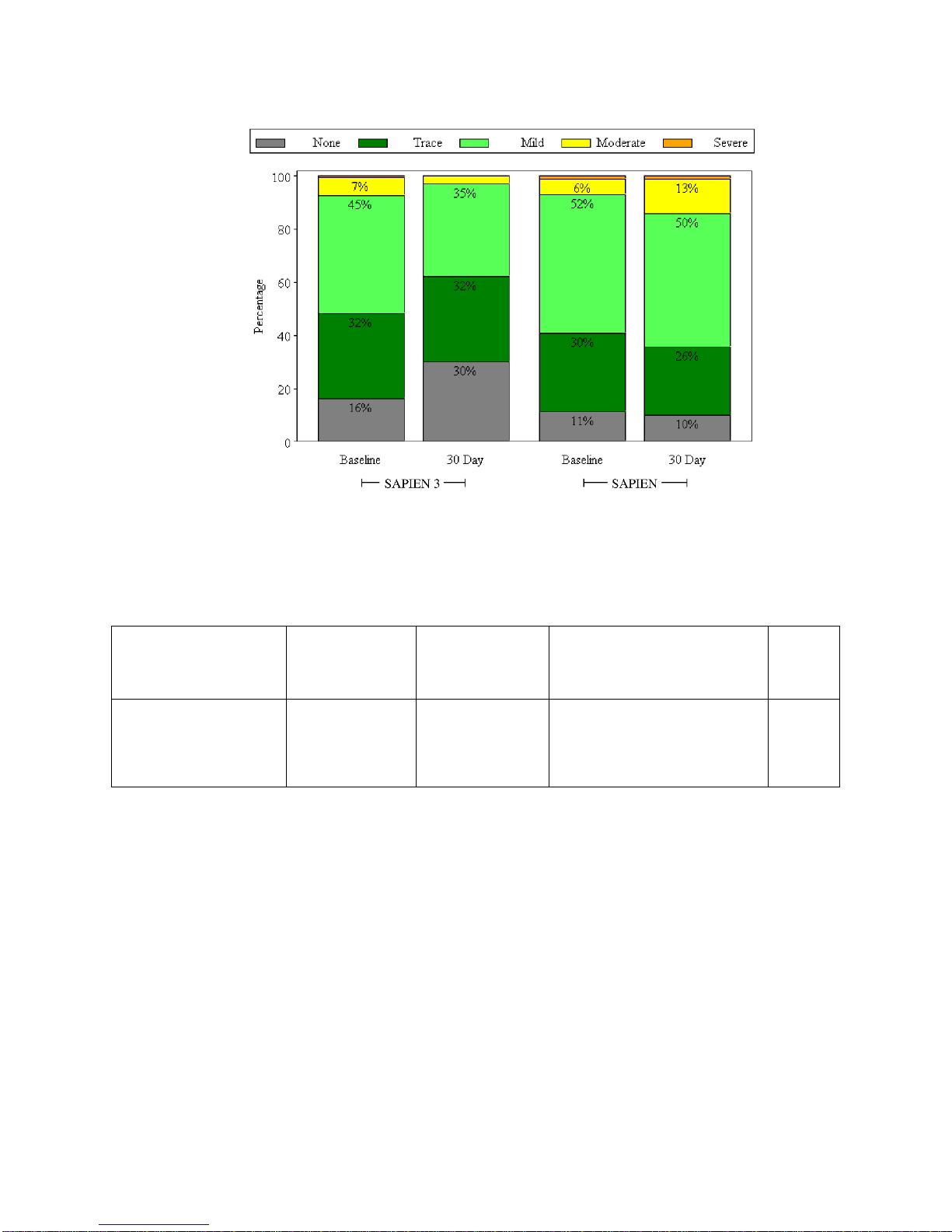
Valve Performance
The mean EOA increased from 0.7 ± 0.2 cm2at baseline to 1.6 ± 0.4 cm2at 30 days, as shown in
Figure 5.
Figure 5:
Effective Orifice Area
(PIIS3HR VI Population)
The average mean gradient decreased from 45.5 ± 14.3 mmHg at baseline to 11.1 ± 4.5 mmHg at 30
days, as shown in Figure 6.
Figure 6:
Mean Gradient
(PIIS3HR VI Population)
Doppler Velocity Index
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Baseline 30 Day
0.7
1.6
Mean Gradient (mmHg)
0
10
20
30
40
50
60
70
80
90
100
110
Baseline 30 Day
45.5
11.1
20
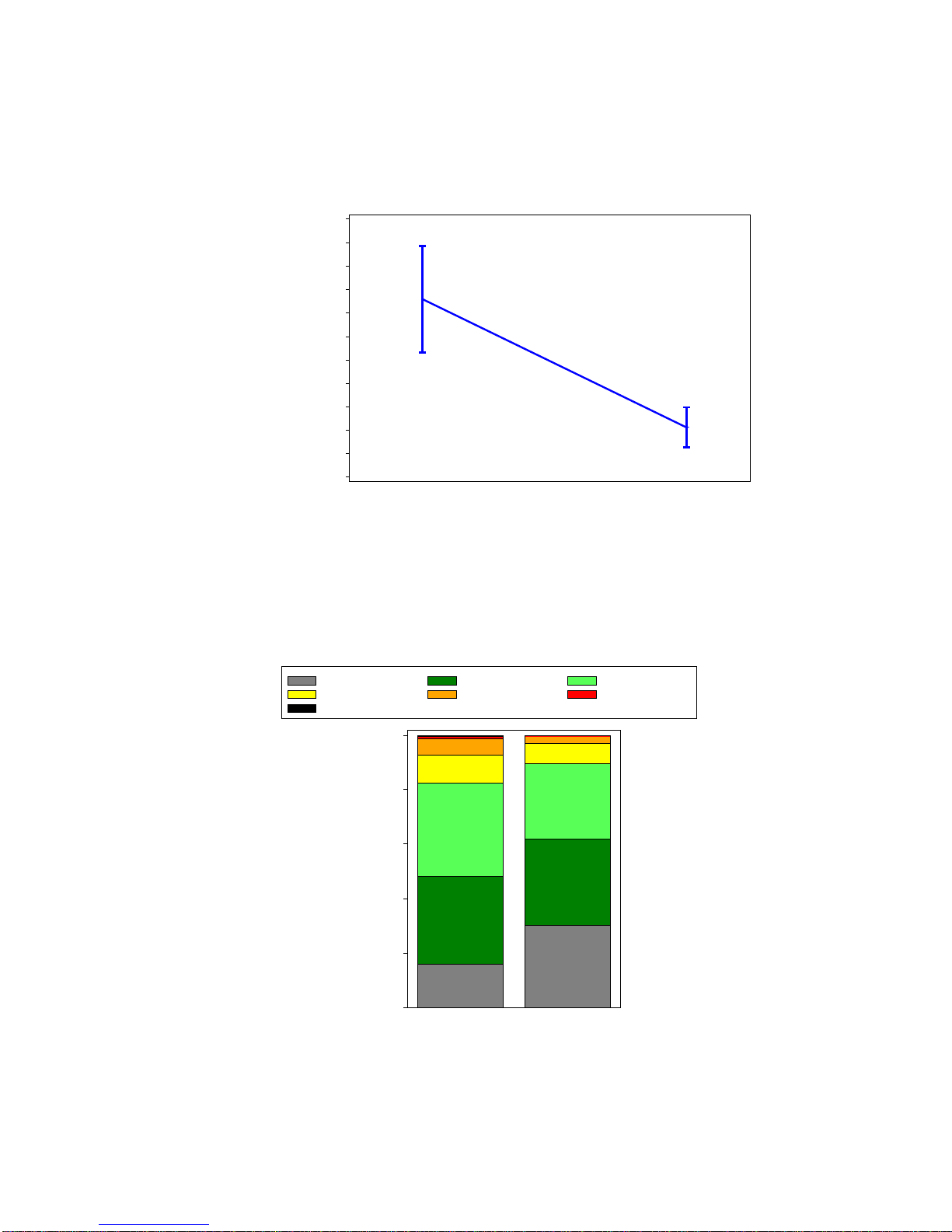
The mean peak gradient decreased from 75.8 ± 22.6 mmHg at baseline to 21.2 ± 8.5 mmHg at 30 days,
as shown in Figure 7.
Figure 7:
Peak Gradient
(PIIS3HR VI Population)
The proportion of patients with AI ≥ moderate was 7.3% at baseline and 3.0% at 30 days, as shown in
Figure 8.
Figure 8:
Aortic Insufficiency
(PIIS3HR VI Population)
Peak Gradient (mmHg)
0
10
20
30
40
50
60
70
80
90
100
110
Baseline 30 Day
75.8
21.2
None Trace Mild
Mild-Moderate Moderate Moderate-Severe
Severe
Percentage
0
20
40
60
80
100
Baseline 30 Day
16%
30%
32%
32%
34% 27%
10% 8%
6%
Table of contents
Other Edwards Medical Equipment manuals

Edwards
Edwards VolumeView EV1000 User manual

Edwards
Edwards SAPIEN XT User manual

Edwards
Edwards SAPIEN XT User manual

Edwards
Edwards ClearSight System User manual

Edwards
Edwards CG16K User manual

Edwards
Edwards EV1000 Clinical Platform User manual

Edwards
Edwards AUTO 306 User manual

Edwards
Edwards EV1000 Clinical Platform User manual

Edwards
Edwards ES Series User manual

Edwards
Edwards TruClip TCLIP05 User manual






















