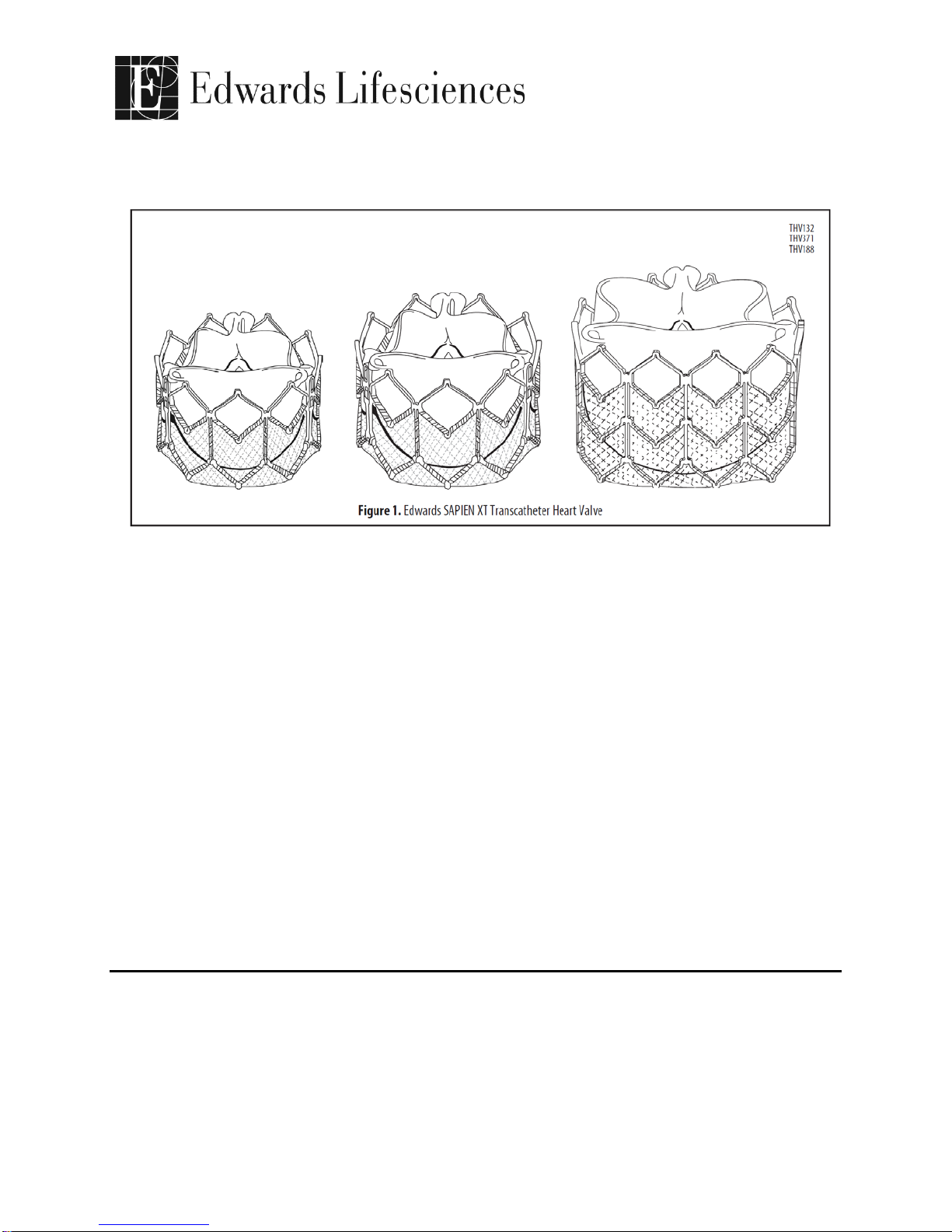
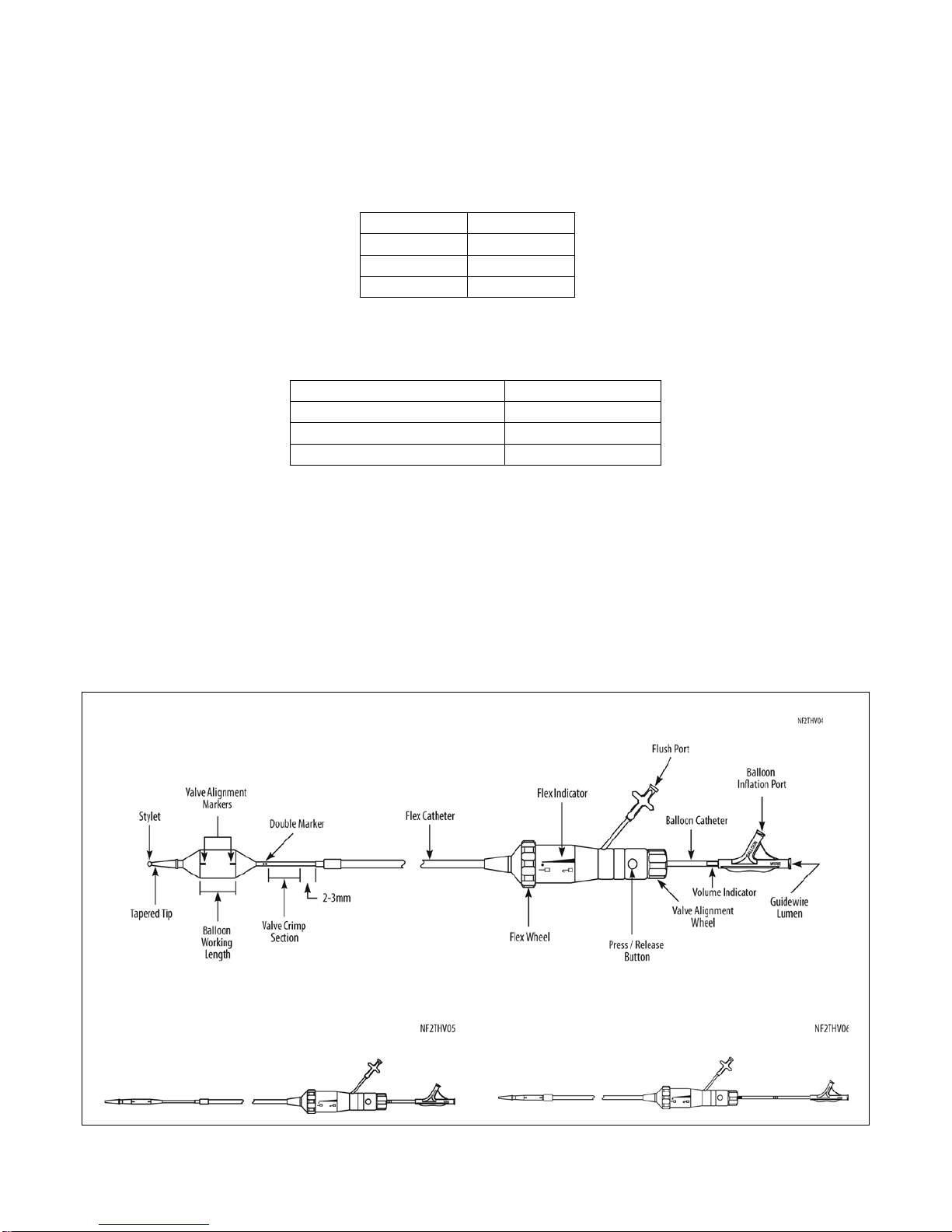
4
•Use of excessive contrast media may lead to renal failure. Measure the patient’s creatinine level
prior to the procedure. Contrast media usage should be monitored.
•Patient injury could occur if the delivery system is not un-flexed prior to removal.
•Care should be exercised in patients with hypersensitivities to cobalt, nickel, chromium,
molybdenum, titanium, manganese, silicon, and/or polymeric materials.
•The procedure should be conducted under fluoroscopic guidance. Some fluoroscopically guided
procedures are associated with a risk of radiation injury to the skin. These injuries may be painful,
disfiguring, and long-lasting.
•THV recipients should be maintained on anticoagulant/antiplatelet therapy as determined by their
physician. This device has not been tested for use without anticoagulation.
•Do not add or apply antibiotics to the storage solution, rinse solutions, or to the THV.
5.0 Precautions
•Safety, effectiveness, and durability of the THV have not been established for implantation within
a previously placed surgical or transcatheter pulmonic valve.
•Long-term durability has not been established for the THV. Regular medical follow-up is advised
to evaluate THV performance.
•Glutaraldehyde may cause irritation of the skin, eyes, nose and throat. Avoid prolonged or
repeated exposure to, or breathing of, the solution. Use only with adequate ventilation. If skin
contact occurs, immediately flush the affected area with water; in the event of contact with eyes,
immediately flush the affected area with water and seek immediate medical attention. For more
information about glutaraldehyde exposure, refer to the Material Safety Data Sheet available from
Edwards Lifesciences.
•Patient anatomy should be evaluated to prevent the risk of access that would preclude the
delivery and deployment of the device.
•To maintain proper valve leaflet coaptation, do not overinflate the deployment balloon.
•Appropriate antibiotic prophylaxis is recommended post-procedure in patients at risk for
prosthetic valve infection and endocarditis.
•Safety and effectiveness have not been established for patients with the following
characteristics/comorbidities:
oEchocardiographic evidence of intracardiac mass, thrombus, or vegetation
oA known hypersensitivity or contraindication to aspirin, heparin or sensitivity to contrast
media, which cannot be adequately premedicated
oPregnancy
oPatients under the age of 10 years
6.0 Potential Adverse Events
Potential risks associated with the overall procedure including potential access complications associated
with standard cardiac catheterization, balloon valvuloplasty, the potential risks of conscious sedation
and/or general anesthesia, and the use of angiography:
•Death
•Respiratory insufficiency or respiratory failure
•Hemorrhage requiring transfusion or intervention
•Cardiovascular injury including perforation or dissection of vessels, ventricle, myocardium or
valvular structures that may require intervention
•Pericardial effusion or cardiac tamponade
•Embolization including air, calcific valve material or thrombus
•Infection including septicemia and endocarditis
•Heart failure
•Myocardial infarction